Contents
- Overview
- Creating a Slurm File
- Running a Slurm File
- Using parallel processing
- Why is my job taking forever?
Overview
For anything that goes beyond running basic lines of code, a job must be submitted to the compute nodes so that the compute nodes can properly handle tasks. Note, this is a requirement on the HPC, as login nodes are not the intended resource for computation. Job submission is handled by the Slurm batch scheduler which allocates compute resources to the job when they become available.
Creating a Slurm File
To create a job, a slurm file must be created:
- Your script (.R, .py, .m, etc.) should already be created (in the
codefolder of your project) and you should have already bug-tested this script on your local machine. Push it to your project repository on GitHub and merge it to themainbranch. - Make sure that you are connected to the FIU VPN using your personal credentials. Follow the instructions here.
- Using Google Chrome as your browser, visit hpcgui.fiu.edu and log in with your personal credentials.
- Under Clusters, select “Panther Shell Access.”
- When prompted, input your password. (It will no look like you are typing anything, but persevere and hit Enter!)
- Navigate, using
cdcommands, to the folder where your script lives. This is where you will want to place the slurm file. - If a nightly sync has not already taken place, manually sync this folder to the GitHub remote with the command
git pull. - Create and open a new file with the command
nano USEFULNAME.subwhere USEFULNAME is replaced by an actually useful name that describes what this .sub file will do. - Type or paste into the empty file the necessary script information. See below for an example that can be customized.
- Save your new file by holding down the ctrl key and pressing the letter ‘o’. (Note: this is ctrl on both PC and Mac, not cmd.) You will be prompted to save the file, which you do by hitting enter/return.
- To exit the nano view, hold down the ctrl key again and press the letter ‘x’.
- Use
lsto inspect the contents of the folder. You should see your new .sub file.
The file below represents a sample Slurm script where a conda base environment is being activated:
#!/bin/bash
#SBATCH --job-name=myjob # job name
#SBATCH --nodes=1 # node count
#SBATCH --ntasks=1 # total number of tasks across all nodes
#SBATCH --time=00:10:00 # total run time limit (HH:MM:SS)
#SBATCH --mem=10G # real memory per node (use G for gigabytes, M for megabytes)
#SBATCH --mail-type=end # send email when job ends
#SBATCH --mail-user=email@fiu.edu # email address
(if running a highmem job:)
#SBATCH --account=iacc_gbuzzell # SLURM account name (delete these 3 lines if not running a highmem job)
#SBATCH --partition=highmem1 # partition name (use high memory nodes)
#SBATCH --qos=highmem1 # QOS
module load singularity-3.8.2
singularity exec --bind /home/data/NDClab /home/data/NDClab/tools/containers/python-3.8/python-3.8.simg python3 filename.py
https://slurm.schedmd.com/sbatch.html
Here is a guide to what these details in the .sub file mean and how they should be customized:
| variable/text | description | how to customize |
|---|---|---|
| #!/bin/bash | tells the HPC to read what follows in the Bash language | Do not modify. |
| job-name | name for the job that displays in squeue | Replace “myjob” with a short, informative name for the job you are running. You can then see this easily on the squeue. |
| nodes | tells the HPC how many nodes (at minimum) should be utilized (nodes are collections of CPUs) | Keep at “1” unless you are doing parallel processing. |
| tasks | tells the HPC how many tasks (that is, CPUs) should be utilized across the number of nodes selected | Keep at “1” unless you are doing parallel processing. |
| time | limits the amount of time the HPC should devote to running the script | Ballpark the time your script will need to run and give yourself some buffer. Please don’t go way overboard and burn through excessive lab time on the HPC. Jobs are killed once walltime is reached. |
| mem | how much memory to allocate to the job (per node) | You don’t want to request way more memory than you need, but you also don’t want to run out of memory, so it may take a bit of trial and error to figure out how much to request here. No need to request 30 GB when you only need 1! If your job errors due to not having enough memory, try raising this value and trying again. Maximum on default nodes is 30 GB (highmem nodes is much higher ~500GB). |
| account | The HPC account used to run the script | By default this will be “acc_gbuzzell” and this line can be omitted. For high memory jobs, the paid queue “iacc_gbuzzell” should be used (along with partition and qos lines specifying the “highmem1” partition). |
| partition | The node partition desired | (see above) defaults are “default-partition” or “default-alternate”, high memory nodes are in the “highmem1” partition |
| qos | quality-of-service | (see above) |
| mail-type | tells the HPC to send you an e-mail when the job is done | If the job is very small and will run quickly, you can delete this line to avoid an unnecessary e-mail in your inbox. Otherwise, leave it unchanged. |
| module load singularity-3.8.2 | loads the IRCC-managed singularity image | Do not modify. (However, if you become aware of a newer image, please PR a suggested wiki update to the lab technician!) |
| singularity exec –bind /home/data/NDClab | binds data within the NDCLab folder to the singularity image | Do not modify. |
| /home/data/NDClab/tools/containers/python-3.8/python-3.8.simg | identity of the selected container | Use the example for Python. For R, use /home/data/NDClab/tools/containers/R/R-4.2.2A/R-4.2.2A.simg |
| python3 filename.py | the actual command to run the script specified | Specify python3 for .py or Rscript for .R. |
MATLAB is a little special. To create a Slurm script for a .m file, replace the singularity line in the sample above with:
module load matlab-2023b
matlab -nodisplay -nosplash -r filename
…where filename is the name of your MATLAB script, but without the final .m.
Running a Slurm File
To run an existing slurm file, you will log into the HPC and utilize the shell interface to execute the script.
- First, make sure that you are connected to the FIU VPN using your personal credentials. Follow the instructions here.
- Using Google Chrome as your browser, visit hpcgui.fiu.edu and log in with your personal credentials.
- Under Clusters, select “Panther Shell Access.”
- When prompted, input your password. (It will no look like you are typing anything, but persevere and hit Enter!)
- Navigate to the folder containing the slurm file (file extension
.sub) that you want to execute usingcdcommands. - Run the script by typing the following into the shell:
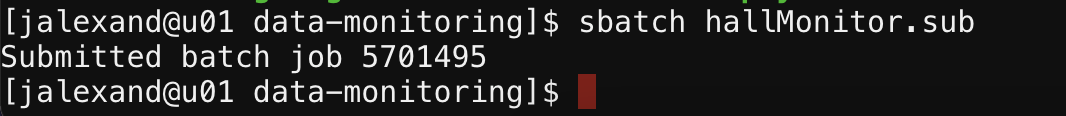
sbatch <filename.sub>. For instance, to execute a hallMonitor.sub file, you would typesbatch hallMonitor.sub. If you want to call your script with different or additional parameters than what is specified in the script with the #SBATCH lines, here is where you need to add those flags and arguments (e.g.sbatch --mem=1000 <filename.sub>, check outsbatch --helpfor info on how to use sbatch effectively). - The shell will output a message to indicate that the batch was submitted and its associated job number:
- Alternatively, the parameters in the “#SBATCH” lines above can be specified on the command line and will override whatever is in the .sub file.
sbatch --mem=3G <filename.sub>will request 3 GB memory for example.
- Alternatively, the parameters in the “#SBATCH” lines above can be specified on the command line and will override whatever is in the .sub file.
- Check status and runtime of currently running scripts with the squeue command:
squeue -u <username>. - Use
lsto see if the process is complete. When it is complete, you will see an output file in the same folder whose name is “slurm-NUMBER.out”:
- Use
cat slurm-NUMBERS.outto print all messages to the console. - If your script is designed to output any new files (for example, a CSV), and assuming you don’t get any error messages, that file should now exist in the appropriate folder.
Using parallel processing
One advantage to submitting jobs to the batch scheduler, instead of just running a script in a HPC terminal or on your local machine, is that you can request multiple cores from Slurm and utilize parallel processing in your job. Running parts of your script in parallel can save time and computing power and minimize the number of jobs you need to request. For example, if you need to process 20 files, rather than submitting 20 separate jobs to the batch scheduler, or running them all sequentially and requesting a large walltime, you can run them in parallel by requesting multiple CPUs and processing multiple subjects at once.
Change the “ntasks” line from 1 to the number of cores you want to request, and make sure your “mem” requested is large enough to handle these multiple processes running concurrently.
Instead of “–mem” you can use “–mem-per-cpu” if you want to specify the memory needed for each worker (mem-per-cpu * # cores = total mem).
In Matlab
You can modify your matlab script to run processes in parallel by adding the lines below:
cluster = parcluster('local');
numCores = maxNumCompThreads; % this should be the maximum cores available to you, same as ntasks * cpus-per-task
parpool(cluster, numCores)
%% And then replace a for loop with a parfor loop
parfor data = 1:length(dataList)
% process data in parallel
end
Keep in mind using parpool in Matlab, that workers must be on the same node. So running on a medium memory node can use at maximum 16 cores (though high memory nodes have from ~40-88 CPUs).
(If you really want to utilize workers across different nodes the “cpus-per-task” option can be used instead of/in conjuction with “ntasks” https://www.mathworks.com/matlabcentral/answers/747522-hpc-slurm-ntasks-and-matlab-parcluster-numworkers-question but aside from this use case ntasks and cpus-per-task should behave the same.)
In Python
You can utilize parallel processing in Python with the “multiprocessing” module.
https://www.sitepoint.com/python-multiprocessing-parallel-programming/
Why is my job taking forever?
The batch scheduler submits jobs to the HPC based on the order in which they’re received, as well as the requested memory allocated, requested walltime, and how many other jobs are currently running. To check on the status of your jobs you can use squeue, squeue -u <username>, where you will see your submitted jobs along with how long they’ve been running and their status (PD=pending, R=running).
If your jobs are sitting in the queue for longer than you would like, you can check on how busy the HPC is with the sar command. A %idle of ~60 or less indicates that the HPC is very busy and may not get to your jobs for a while, especially if they’re large memory or long walltime jobs. If you think your job can afford to be submitted with less memory or walltime, you can try cancelling scancel NUMBERS and resubmitting the job with fewer resources requested.